Forekomst og arvelighed af Huntingtons sygdom
Registreringen foregår i det danske Huntingtonregister, der vedligeholdes af en forskningsgruppe under Institut for Cellulær og Molekylær Medicin ved Københavns Universitet. Registeret rummede i 2001 318 slægter, hvoraf nogle er kortlagt tilbage til starten af 1800-tallet.
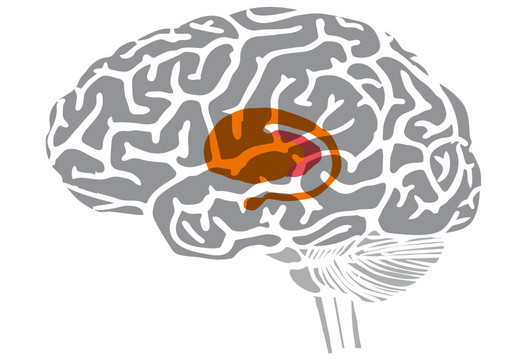
Ved Huntingtons sygdom ses en gradvis degeneration af neuronerne først i striatum – især i nucleus caudatus og putamen – og siden også i hjernebarken. Huntingtons sygdom påvirker især den dybtliggende hjernestruktur striatum (nucleus caudatus og putamen), her markeret med orange.
Arvelighed
Huntingtons sygdom er en dominant arvelig sygdom, der skyldes en mutation i HD-genet på kromosom 4.
Sygdommen optræder således i familier, rammer lige mange mænd og kvinder og overføres fra generation til generation uden overspring. Hvis man er bærer af det genetiske anlæg for Huntingtons sygdom vil man, hvis man lever længe nok, uundgåeligt udvikle sygdommen.
Får man børn, vil de have 50 % risiko for at arve sygdommen. Har man ikke det genetiske anlæg for Huntingtons sygdom, kan man hverken få sygdommen eller give den videre til sine børn.
Ved hjælp af en blodprøve kan det afklares, om man er bærer af det genetiske anlæg eller ej. Sikkerheden af svaret er meget tæt på 100 %. Også ved fosterundersøgelse kan sygdommen påvises. Genetisk testning af 'raske', symptomfri personer rejser imidlertid en række etiske og praktiske problemstillinger.
HD-genet
HD-genet koder for proteinet huntingtin, hvis normale funktion ikke kendes fuldt ud. Personer uden Huntingtons sygdom har mellem 6 og 35 gentagelser (repeats) af DNA-sekvenser med baserne CAG (cytosin-adenin-guanin) på genet, hvorimod bærere af sygdommen har mindst 36 gentagelser. 36-39 gentagelser udgør en slags gråzone, men ved 40 gentagelser eller mere ses fuld penetrans.
Debutalderen for Huntingtons sygdom er omvendt proportional med antallet af repeats. CAG-repeat-ekspansionen er ustabil mellem generationer. Store ekspansioner sker typisk i den mandlige meiose (dannelsen af kønsceller).
Patienter, der udvikler Huntingtons sygdom i barndommen, har næsten altid arvet anlægget fra faderen. Huntingtons sygdom er således genetisk set mere uensartet end mange andre neurodegenerative sygdomme.
